Overview
About CCR
The Center for Cancer Research (CCR) is home to approximately 250 Principal Investigators and Senior Investigators, of which one third are located in Frederick. CCR is organized into over 50 branches and laboratories, each one grouping scientists with complementary interests. CCR’s investigators are basic, clinical, and translational scientists who work together to advance our knowledge of cancer and AIDS and to develop new therapies against these diseases. CCR investigators collaborate with scientists at the more than 20 other Institutes and Centers of the National Institutes of Health (NIH), as well as with extramural scientists in academia and industry.

Optical Microscopy
The optical microscope, often referred to as the “light microscope”, is a type of microscope which uses visible light and a system of lenses to magnify images of small samples. The image from an optical microscope can be captured by normal light-sensitive cameras to generate a micrograph.
Quick Facts
NCI Optical Microscopy labs utilize basic and advanced microscopy techniques
Five different laboratory sites are available in Bethesda and three in Frederick
Please visit each laboratory for contact information
Scheduling time on instruments is on a per core/per instrument basis
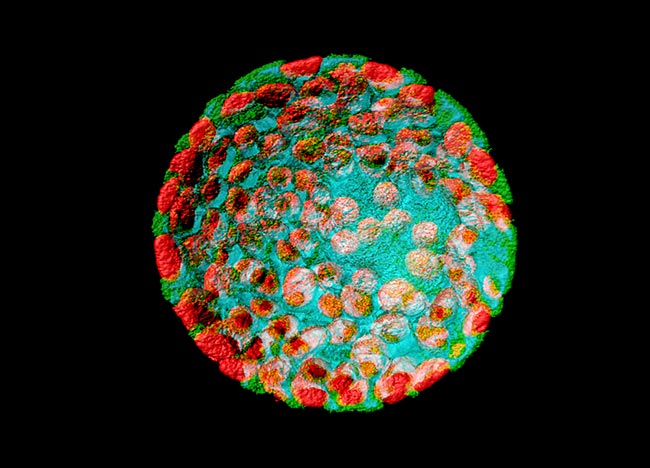
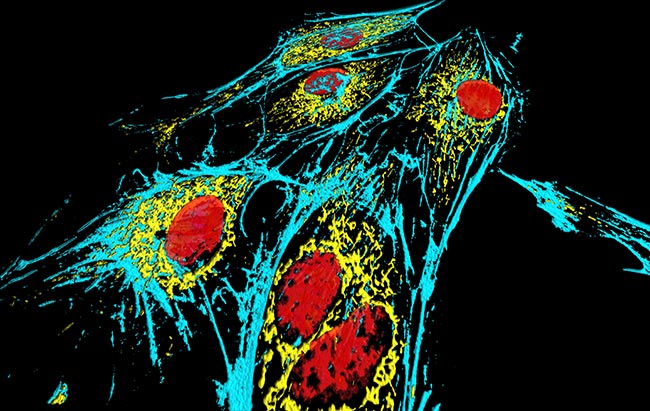
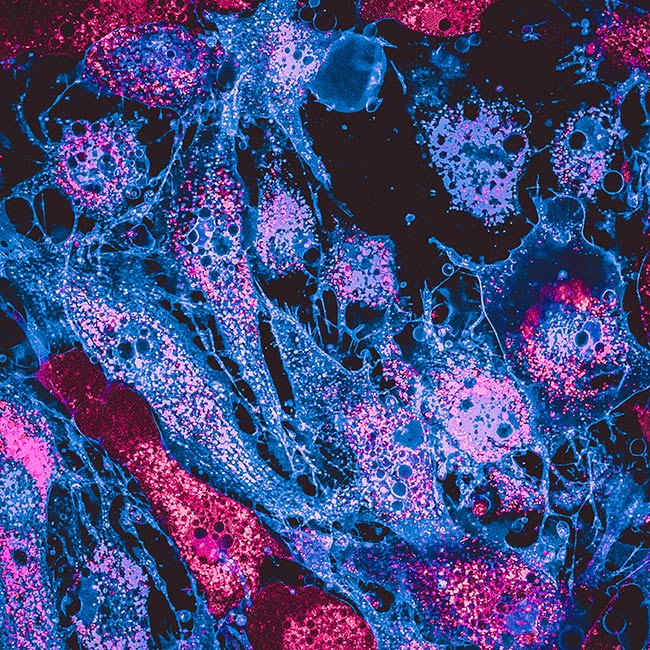
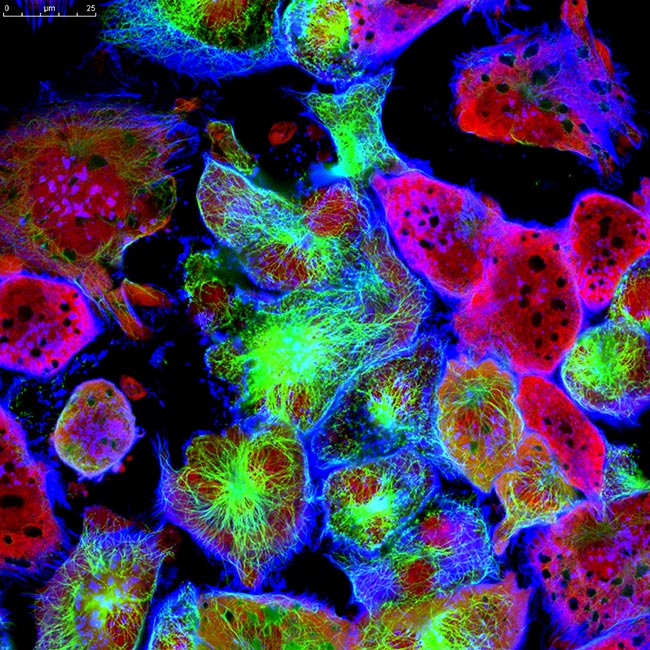
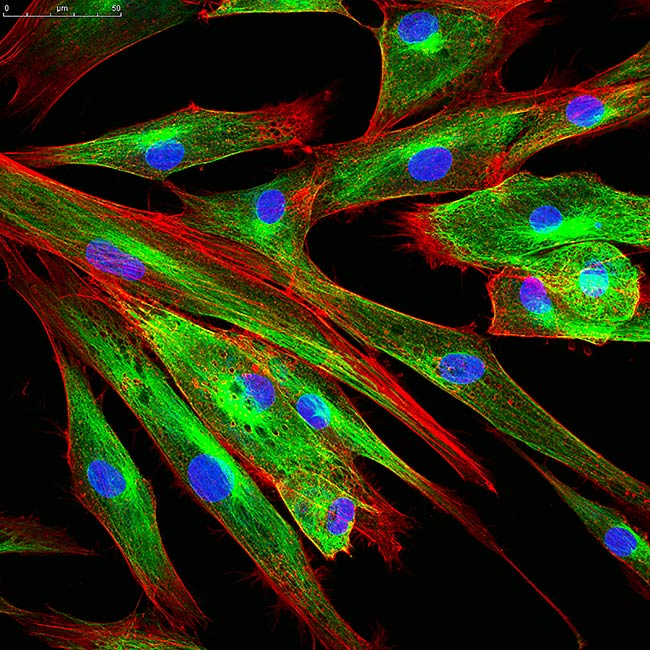
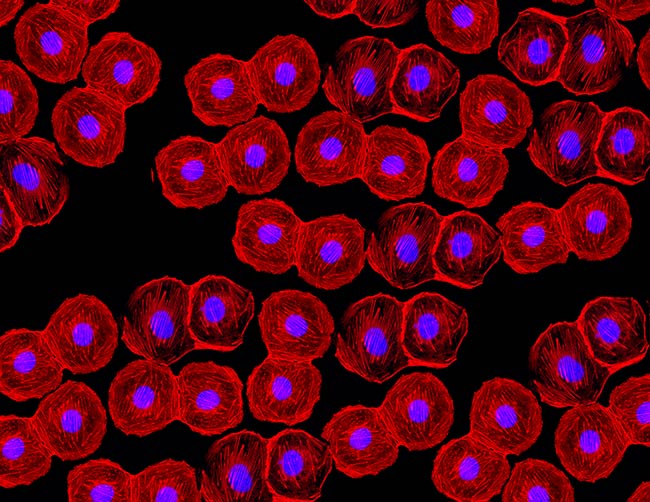
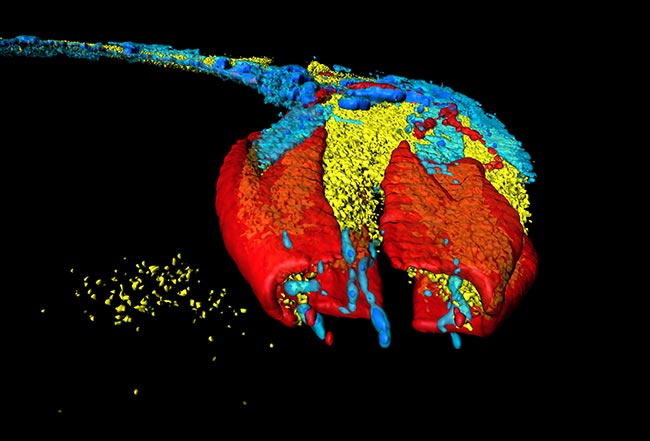
Representative Images From the Cores
NCI Optical Microscopy produces exceptional imagery. Browse the image gallery to see more.

Building 37, Room 2066
37 Convent Drive
Bethesda, MD 20892
Building 37, Room B114E
37 Convent Drive
Bethesda, MD 20892
Building 37, Room 1066
37 Convent Drive
Bethesda, MD 20892
Building 41, Room C615
10 Center Drive
Bethesda, MD 20892